
Computational Biology Lab @ Duke

Yi Zhang, Ph.D.
Yi is an Assistant Professor at Duke University in the Department of Neurosurgery and Department of Biostatistics and Bioinformatics. Yi obtained PhD in Bioengineering with Dr. Jun S. Song at University of Illinois at Urbana-Champaign. She joined Dr. X. Shirley Liu’s lab and with co-mentors Dr. Myles Brown and Dr. Xihong Lin, as a Research Fellow at Department of Data Science, Dana-Farber Cancer Institute and Harvard University School of Public Health. She has been developing machine learning methods that leverage large-scale single-cell datasets to understand cell states in tissue microenvironment, and also machine learning/AI methods for high-resolution spatial transcriptomics data. She has also developed integrative genomic methods to identify functional gene regulatory mechanisms behind disease-associated human genetic variants. Ongoing work, funded by NIH NIGMS R35 Award and Duke Brain Tumor Center, includes statistical and deep learning modeling of single-cell and spatial transcriptomics and computational pathology, to understand any heterogenous & complex tissues and diseases .
- We are open in recruiting postdocs, students, and staff. Interested in computational biology, ML/AI, single-cell multi-omics, spatial transcriptomics, computational pathology, tumor immunology, or brain? - Welcome to join us!
- We lead development of computational, statistical, and ML/AI methods that leverage large-scale and multi-omic data to understand cancer initiation, progression, and therapeutic responses.
- We are interested in disease systems of multi-cellular components.
- We foster an interdisciplinary, inclusive, and supportive environment. We welcome computational biologists, bioinformaticians, computer scientists, MDs, immunologists, and oncologists.
- We are enthuastic collaborators to work on real-world biological and translational genomics problems.
- Postdocs or PhD soon-to-be who work on areas like computational biology, bioinformatics, single-cell multi-omics, new spatial transcriptomics, deep learning models, genetic variant function, and biological questions in cancer or tumor microenvironment - Welcome to contact and join us! If possible please include (1) CV or Resume (2) A short research statement describing previous/ongoing work and proposed research and interest (3) 1-3 representative publications (published, accepted, or preprint) (4) Three names of references and contact information.
- Prospective students interested in our research are encouraged to apply through Duke graduate programs: Computational Biology and Bioinformatics (CBB - PhD), Biostatsistics (PhD & MS), UPGG (PhD). Graduate admission decisions are made by program committee. MS students from Biostatistics, Biomedical Engineering, Computer Science, ECE etc. are welcome for research interns/assistantships. If contacting for interest, please include (1) CV or Resume and if available (2) Short description of research experience and interest (3) Three names of references. Thanks!
Research Interests
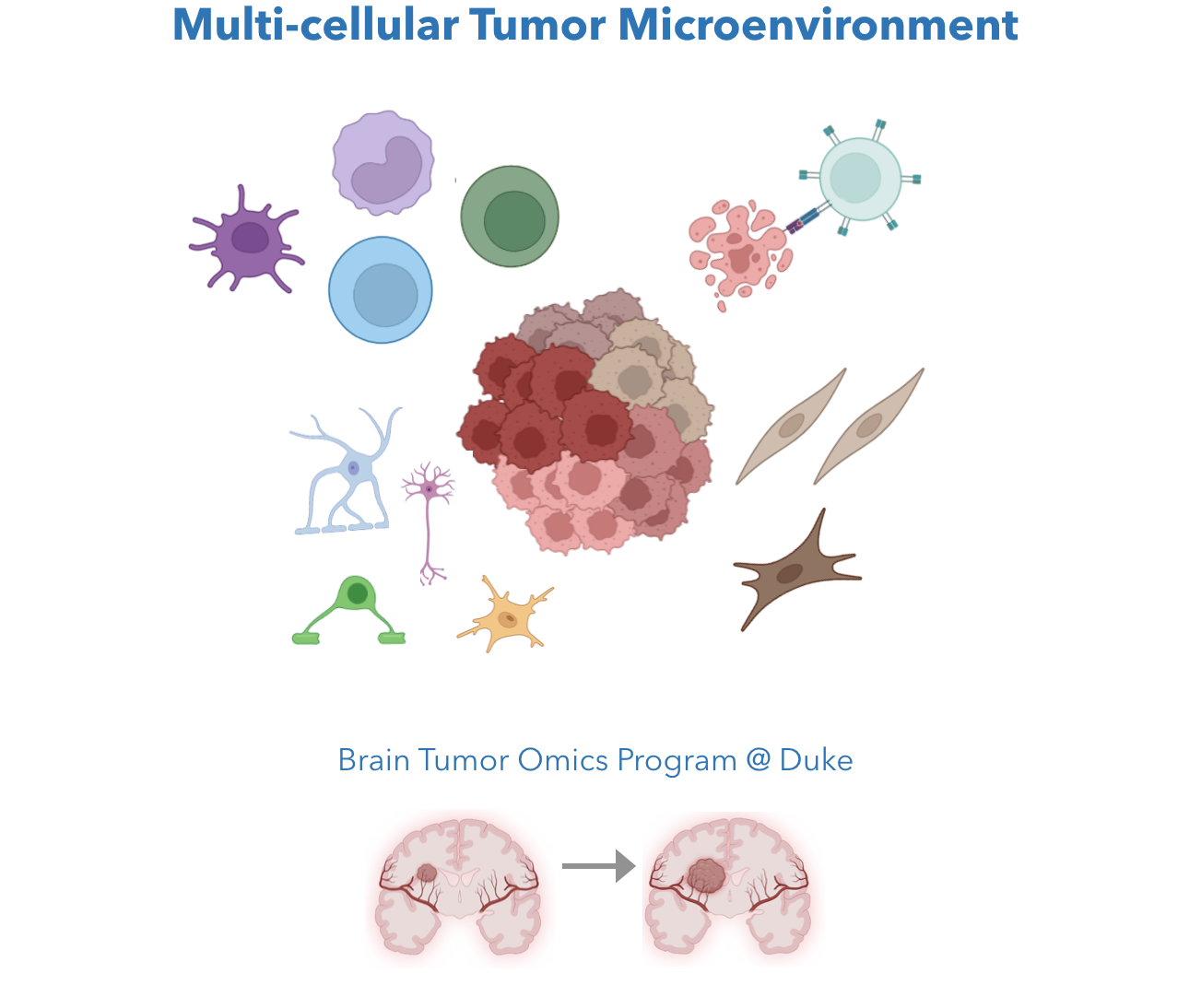
Tumor, like many multi-cellular disease systems, are composed of multiple cell types. For solid tumor, the Cancer-intrinsic ("Seed") properties like proliferation, mutations, and epigenetic changes, and the Cancer-extrinsic ("Soil") properties like tumor-infiltrating immune cell states and inflammation, both affect tumor progression and therapeutic responses. We aim to understand molecular gene programs of tumor microenvironment (TME) cell states, pinpoint functional cancer-TME interactions, and identity targetable tumor immunity modulators.
We are core members of the Brain Tumor Omics Program (BTOP) at Duke Preston Robert Tisch Brain Tumor Center. We will use our computational expertise to develop methods that allow us to understand the incurable tumor types and to improve cancer therapy efficacy.
Related work: MetaTiME for TME cell states ( Zhang et al. Nature communications 2023); Low-grade glioma variant (Manjunath et al. Neuro-Oncology, Song Lab, UIUC). Ongoing: (1) Brain & Breast tumor intrinsic and extrinsic cell states; (2) Tumor-infiltrating macrophage states; (3) Spatial transcriptomics for brain tumor.
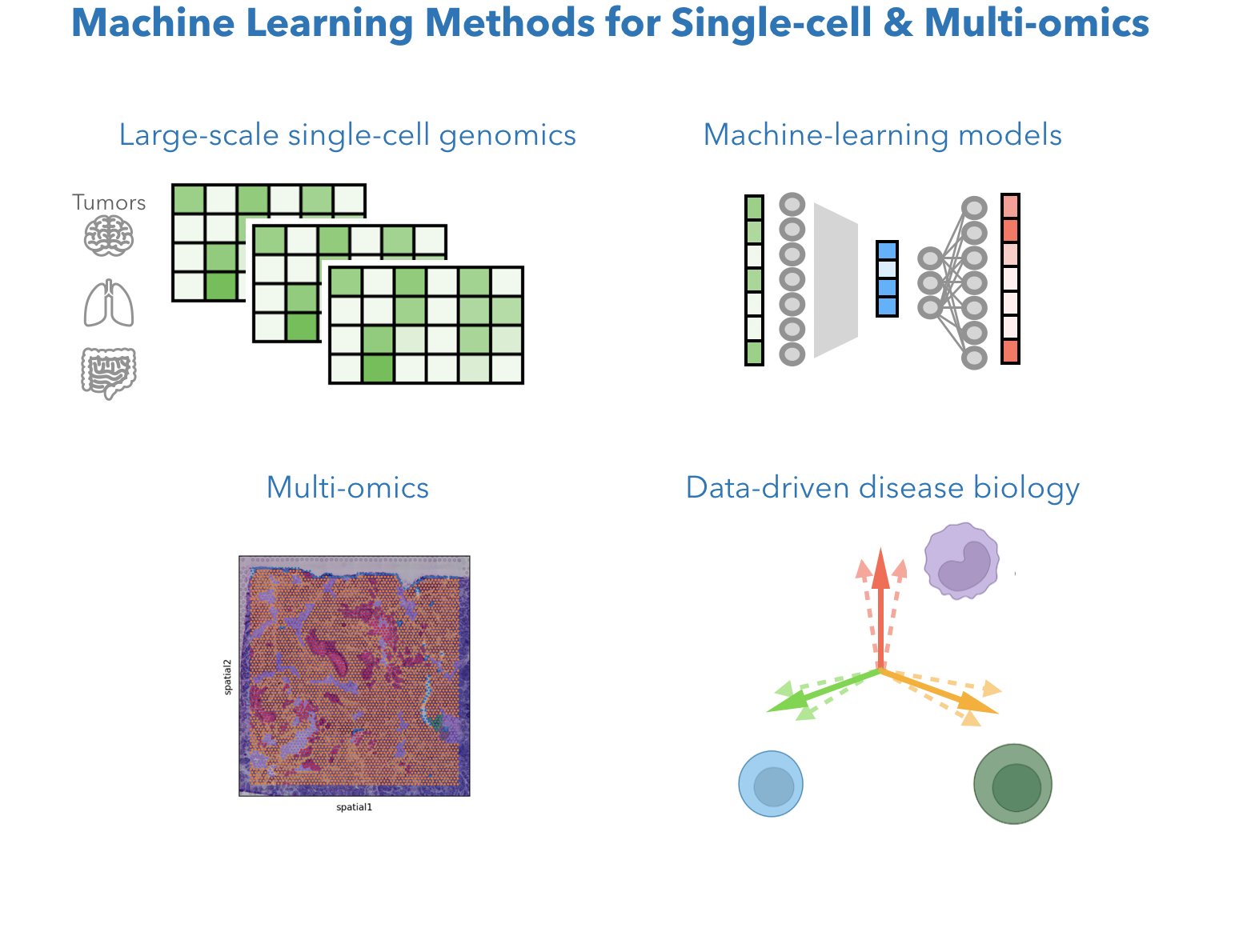
Single-cell genomics data provide high-dimensional measurement of gene expression for each cell. How do we mathematically describe a cell? How do we find cell types or cell states most relavant to biology? How do we integrate the power of the large number of cell data points to understand what roles each cells are playing in tumors? We develop computational methods leveraging machine learning models, large-scale single-cell genomics data, and multi-omic datatypes like spatial transcriptomics and multiome. Currently, we are particular interested in low-dimensional methods and deep learning models with interpretability, as well as image AI models integrated with genomics ML models.
Related work: STHD: machine learning model for high-resolution spatial transcriptomics (Sun* and Zhang#, BioRxiv 2024), *Software: STHD for VisiumHD. MetaTiME for TME cell states ( Zhang et al. Nature communications 2023). Software: MetaTiME cell state annotator toolkit, Cross-cohort training pipeline. Ongoing: (1) Machine learning method for large-scale single-cell transcriptomics integration & interpretation; (2) Timecourse single-cell multiome (ATAC+RNA) modeling; (3) Spatial transcriptomics.
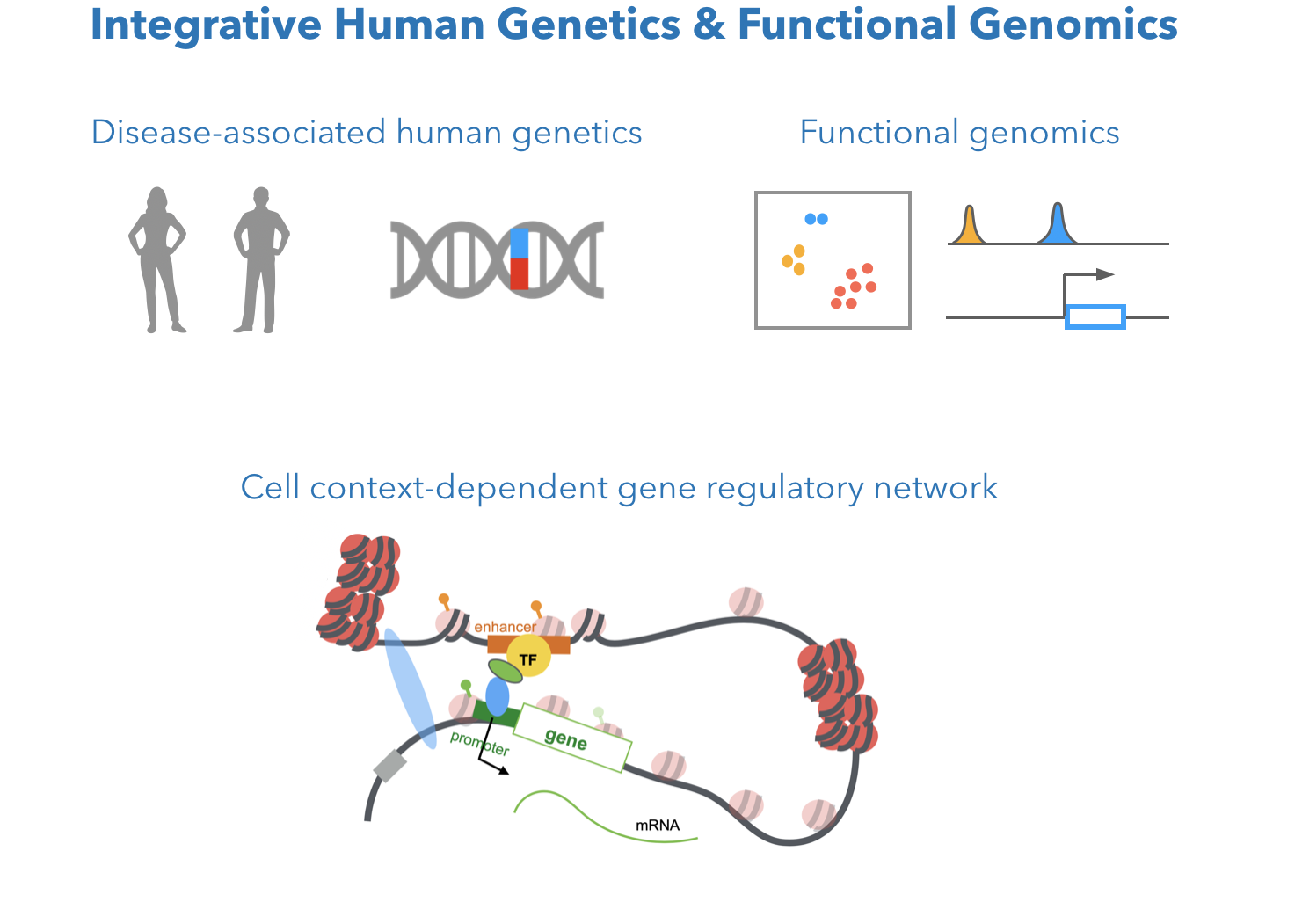
Human genetic variants are natural probes to investigate cell context-dependent gene regulation related to human disease. Genome-wide association studies (GWAS) identified many genetic variants associated with cancer susceptibility. What are the molecular mechanisms behind these associations? What cell types mediate gene modulation for a certain disease trait? Our previous computational methods integrated multiple NGS data (Zhang et al., 2018 Bioinformatics), and integrated eQTL, haplotype-specific expression, epigenetics, motif simulation, and 3D chromatin interactions, to identify gene regulatory effect of functional variants in enhancers (Gene regulatory non-coding variant, Zhang et al. 2018 Cancer Research). We were among the first to infer cell-dependent effect of variant-associated gene regulation (Breast variant and immune cell, Zhang et al. 2019 Frontiers in Genetics). We are interested in further investigate the cell-dependent effect of genetic variants by marrying GWAS summary statistics and functional genomics in ongoing work.